医療関係者の皆さま
医療関係者の皆さまへ
国内の医療関係者(医師、薬剤師、その他の医療関係者)を対象に、
アミカス・セラピューティクス株式会社の医薬品を適正にご使用いただくための情報をご提供します。
日本人を含む遅発型ポンペ病を有する成人患者を対象とした
二重盲検無作為化試験
(PROPEL試験、ATB200-03試験、国際共同第3相試験)1)
警告、禁忌を含む注意事項等情報等については、電子化された添付文書(製品DI)をご参照ください。
1) 承認時評価資料(申請時評価資料:ポンペ病患者における臨床試験(ATB200-03)(2025年6月24日承認、CTD2.7.6.4、CTD2.7.3.2.2.3.4))
試験概要
目的
酵素補充療法(ERT)の治療歴を問わない成人遅発型ポンぺ病患者を対象とし、ポムビリティ(遺伝子組換え)とオプフォルダ併用又はアルグルコシダーゼ アルファとプラセボ併用を52週投与し、有効性及び安全性を比較評価
試験デザイン
多国籍(24ヵ国)、多施設共同(62施設)、二重盲検、無作為化、実薬対照(第3相試験)
対象
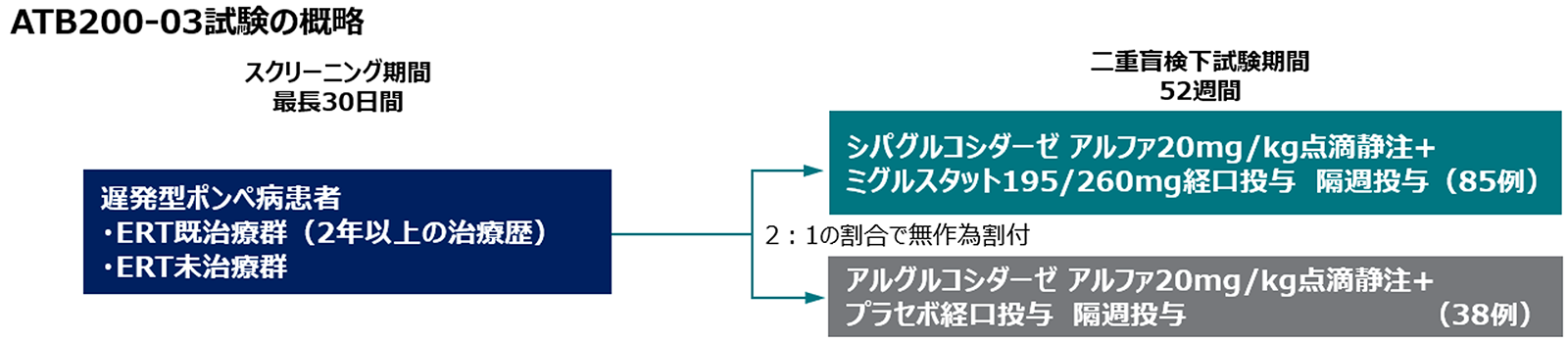
ERTの治療歴を問わない成人遅発型ポンぺ病(LOPD)患者(18歳以上、体重40㎏以上)
試験方法
30日間のスクリーニング期間が設定され、ERT治療歴(ERT既治療又はERT未治療)及びベースライン時の6分間歩行距離のカテゴリー(75m以上150m未満、150m以上400m未満又は400m以上)で層別した上で、ポムビリティとオプフォルダ併用又はアルグルコシダーゼ アルファとプラセボ併用のいずれかに2:1の割合で無作為に割り付け、52週間隔週で投与
評価項目
有効性評価はベースライン時(Day0)及びDay1から12週ごとに実施
主要評価項目は、6分間歩行距離(6MWD)(m)の52週時でのベースラインからの変化量を評価(検証的解析項目)
重要な副次評価項目は、座位努力性肺活量(座位FVC)(予測値に対する%)の52 週でのベースラインからの変化量、徒手筋力検査(MMT)
下肢スコアの52週でのベースラインからの変化量、6MWD(m)の26週でのベースラインからの変化量、PROMIS(身体機能)合計スコアの52週でのベースラインからの変化量、PROMIS(疲労)合計スコアの52週でのベースラインからの変化量、GSGC
合計スコアの52週でのベースラインからの変化量を評価
副次評価項目は、最大吸気圧(MIP)(予測値に対する%)、最大呼気圧(MEP)(予測値に対する%)などを評価
サブグループ解析は、(ERT治療歴ありなし群)による6分間歩行、座位FVC(予測値に対する%)を評価
有害事象などの安全性、免疫原性を評価
解析方法
人口統計学的及びベースライン特性は、Intent-to-Treat(ITT)集団及び安全性解析対象集団で要約し、既往歴、前治療薬及び非薬物療法は安全性解析対象集団で解析した。
有効性の主解析は、有効性の主要評価項目を本併用群と対照薬群の間で、片側有意水準0.025で群間比較することにより実施した。ITT-OBS集団を対象に、有効性の主要評価項目(6MWDの52週でのベースラインからの変化量)を反復測定混合効果モデル(MMRM)により解析し、本併用群とアルグルコシダーゼ アルファ/プラセボ群を比較した。各時点で最小二乗平均値、SE、最小二乗平均値の投与群間差、及び95%CIを推定した。また、解析順序をつけた6つの重要な副次評価項目を設定した。最初に主要評価項目の検定を有意水準0.025で実施し、有意であれば、順位付けした重要な副次評価項目も同様に有意水準0.025で座位努力性肺活量(座位FVC)(予測値に対する%)の52 週でのベースラインからの変化量、徒手筋力検査(MMT) 下肢スコアの52週でのベースラインからの変化量、6MWD(m)の26週でのベースラインからの変化量、PROMIS(身体機能)合計スコアの52週でのベースラインからの変化量、PROMIS(疲労)合計スコアの52週でのベースラインからの変化量、GSGC 合計スコアの52週でのベースラインからの変化量の順に検定を行った。重要な副次評価項目の統計学的有意性は、それぞれ片側有意水準0.025で検定順序に従って解釈した。いずれかの段階で帰無仮説が棄却されなかった場合、それ以降の比較検定は優越性に関して統計学的に有意であると判定しないものとした。重要な副次評価項目は、ITT-LOCF集団を対象に、それぞれANCOVAを用いて解析した。ANCOVAの推定値(各投与群の最小二乗平均値、最小二乗平均値の差、推定値のSE、最小二乗平均値の差の95%CI、及び2群間の比較についてのp値)を示した。
<有効性解析に用いた解析対象集団の定義>
ITT集団:
治験薬を少なくとも1回投与された無作為化された患者全員から構成される。
ITT-OBS集団:
すべての利用可能な観察データを用いて、ベースライン後の欠測データに対する補完を行わないITT集団。つまり、52週を含むいずれの時点の欠測値も補完しない。
ITT-LOCF集団:
欠測データをベースライン後の最後の利用可能なデータで補完したITT集団。
安全性解析は安全性解析対象集団を対象として実施した。有害事象の要約は、投与群別及び投与された患者全体について、患者数と割合を用いて示した。
有効性
主要評価項目:52週時の6MWDの変化量(ITT集団、検証的解析結果)
本併用群は対照薬群に比べて改善が認められたものの、優越性を検証するには至らなかった(p=0.608)。
ITT-OBS集団における52週時の6分間歩行距離のMMRM(Mixed-effect model for repeated measures)から得られたベースラインからの変化量の最小二乗平均値の群間差は5.33m (95%CI: -15.2, 25.9)と本併用群は対照薬群に比べて改善が認められたものの、優越性を検証するには至らなかった(p=0.608、両側p値)。
事前の解析計画にもとづき、正規分布の仮定を含め、6MWDのMMRM解析の仮定を検証したところ、正規分布に従っていないことが確認されたため、治療効果をより適切に推定するために、主要評価項目の代替解析として、ノンパラメトリックrandomization-based ANCOVAを実施し、以降は名目上のp値として解析。
以降の有効性のデータは、ITT集団から外れ値の基準に該当する1例*を含めない場合について実施した解析結果である。
* ERT未治療患者1例において、確実に選択基準を満たして本試験に参加するために、スクリーニング時の6MWT及びPET(肺機能検査)で意図的に低い成績を出していた。
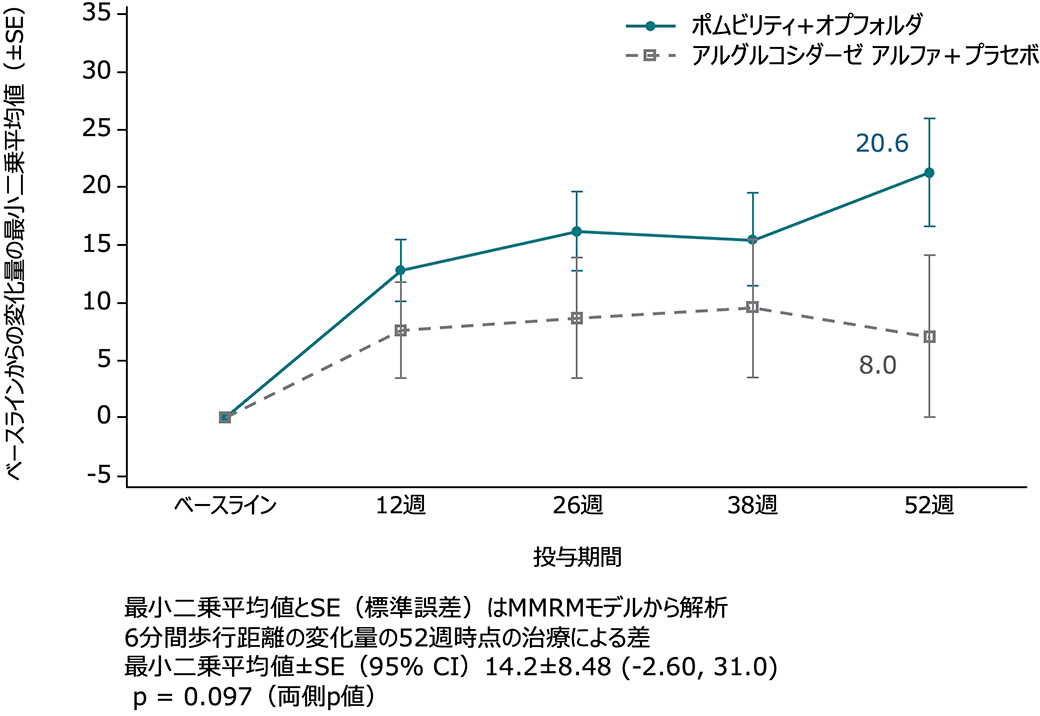
主要評価項目:52週時の6MWDの変化量
本併用群では、52週にわたって変化量の改善状態を示しましたが、対象群と名目上の統計学的優越性は示されなかった(p=0.097)。
6MWDのベースラインからの変化量の推移 (外れ値の患者を除いたITT-OBS集団、名目上のp値)
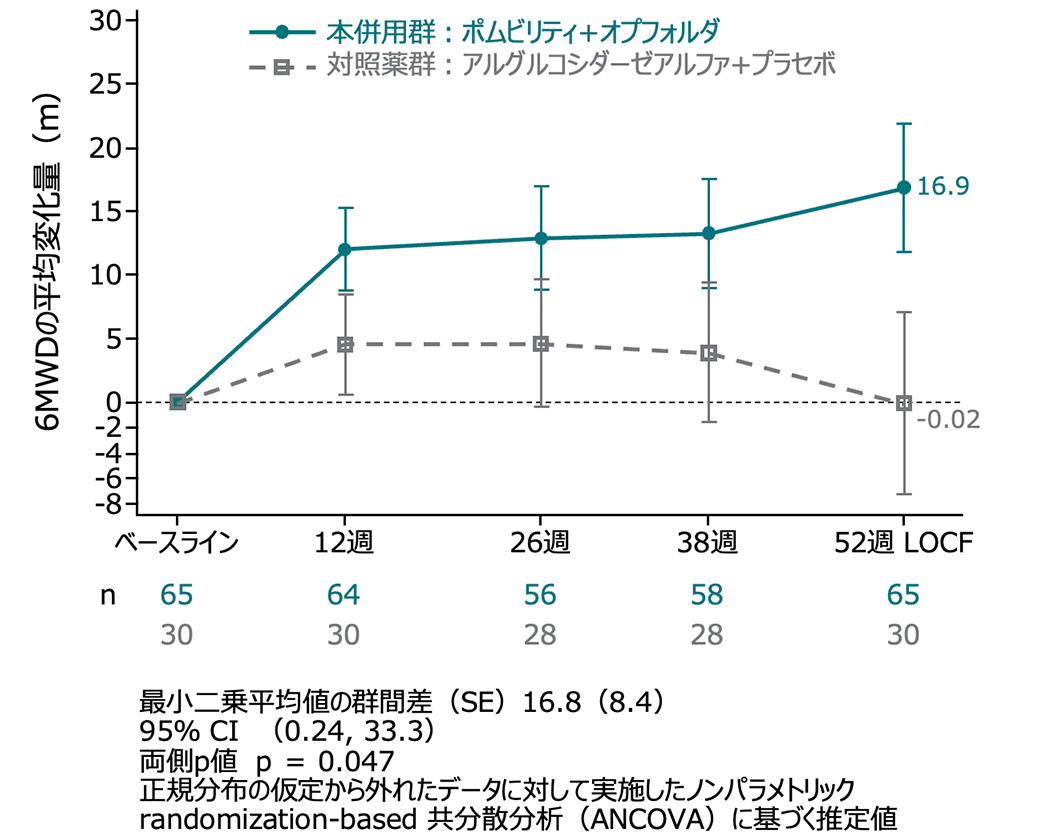
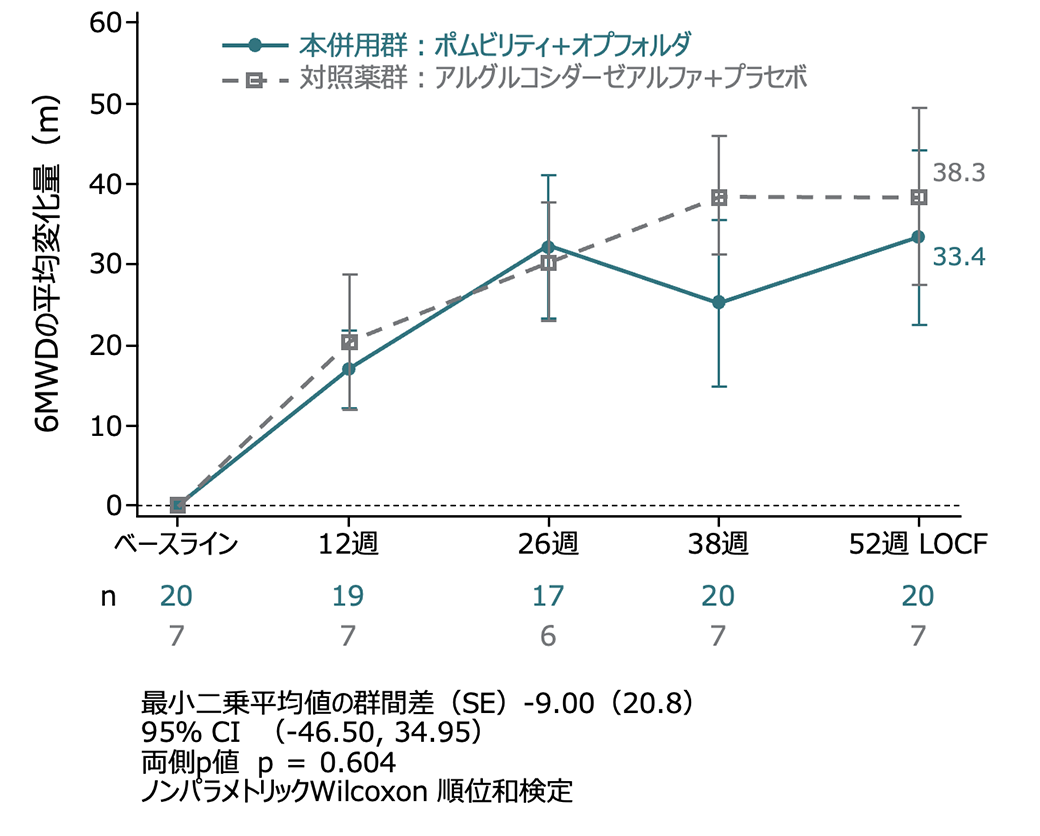
サブグループ解析:6MWD(ERT既治療及びERT未治療群)
ERT既治療患者群は有意な改善がみられ(p=0.047)、ERT未治療群では、有意な差はなかった(p=0.604)。
ERT 既治療患者集団 ベースラインからの変化量
(外れ値の患者を除いたITT-LOCF集団、名目上のp値)
ERT 未治療患者集団 ベースラインからの変化量
(外れ値の患者を除いたITT-LOCF集団、名目上のp値)
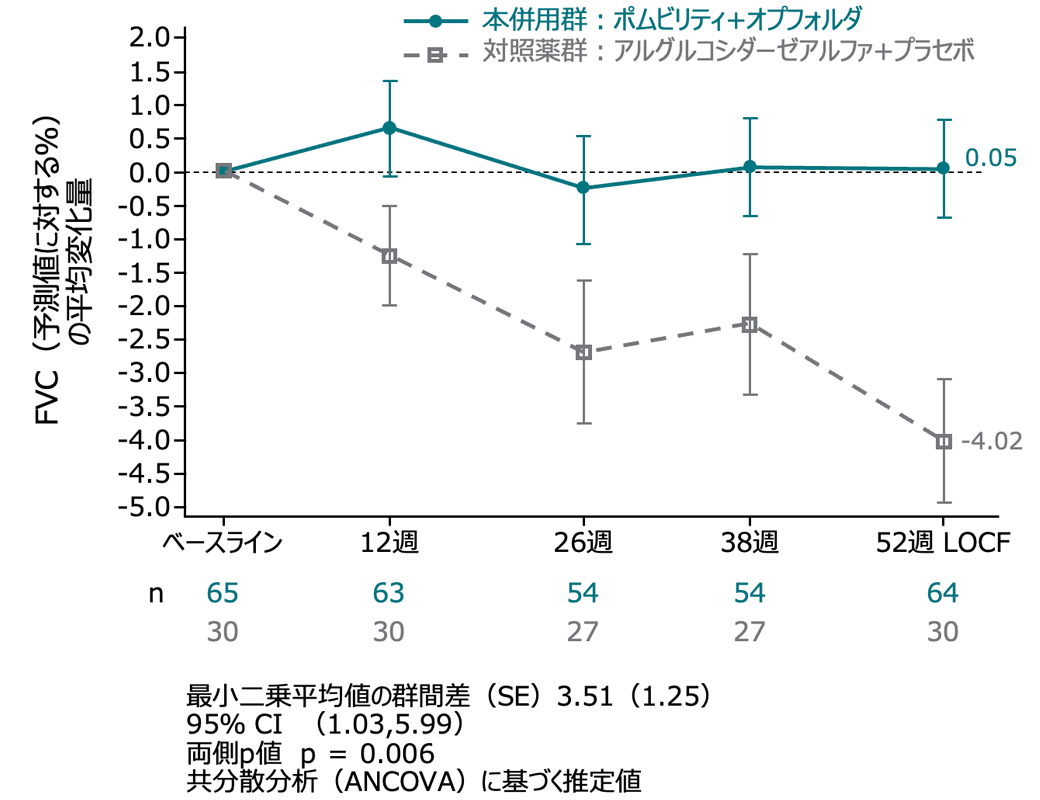
副次評価項目:座位FVC(予測値に対する%)の変化量
本併用群は、対照薬群と比較して座位FVCを統計学的に有意に改善した(p=0.023)。
座位FVC(予測値に対する%)の変化量の推移(外れ値の患者を除いたITT-LOCF集団、名目上のp値)

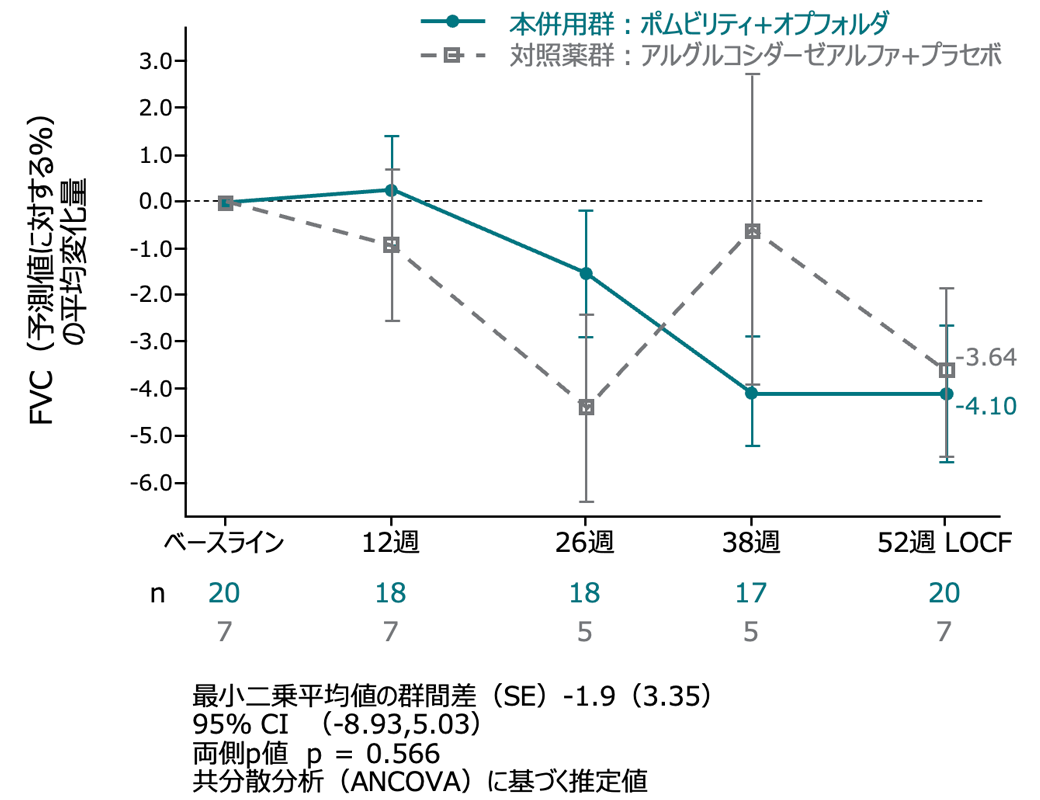
サブグループ解析:座位FVC(予測値に対する%) ERT既治療及びERT未治療群
ERT既治療患者では、52週にわたって横ばいに推移し、対象群と比べて統計学的有意な差を示した(p=0.006)。
ERT未治療患者では、2群間に統計的な有意な差はなかった(p=0.566)。
ERT 既治療患者集団
(外れ値の患者を除いたITT-LOCF集団、名目上のp値)
ERT 未治療患者集団
(外れ値の患者を除いたITT-LOCF集団、名目上のp値)
主要評価項目、重要な副次評価項目及びその他の副次評価項目の要約(外れ値の患者を除く)①
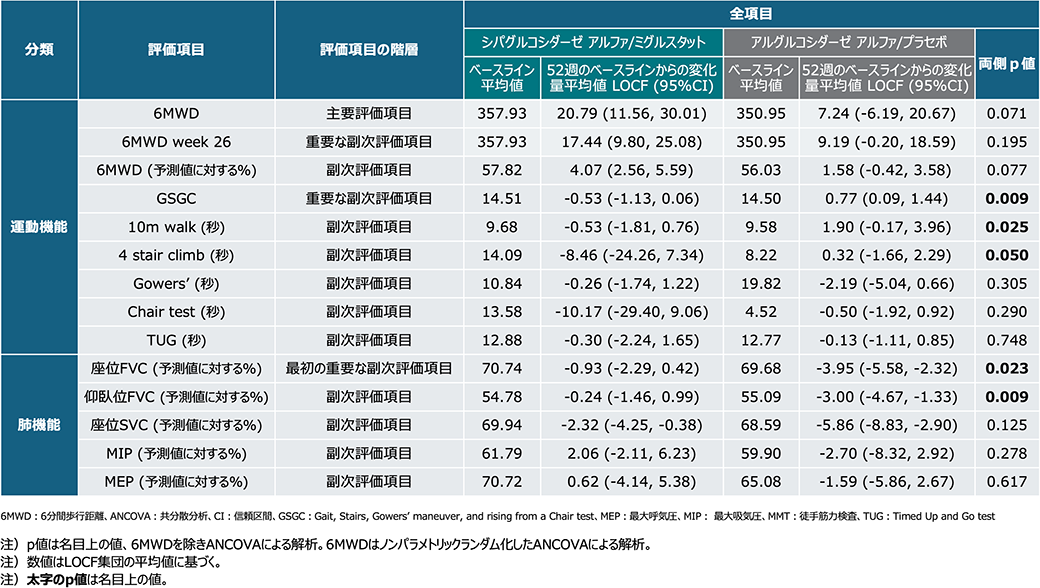
主要評価項目、重要な副次評価項目及びその他の副次評価項目の要約(外れ値の患者を除く)②

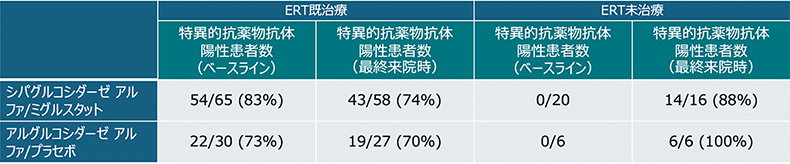
免疫原生
抗薬物抗体陽性者の割合、抗体価と有害事象発現の間には、関連性は認められなかった。
安全性
有害事象
重篤な有害事象は、本併用群が9.4%(8/85例)、対照薬群が2.6%(1/38例)に認められ、このうち治験薬と関連がある有害事象は本併用群1件(アナフィラキシー様反応)であり、当該事象により治験薬の投与は中止された。
治験薬の投与中止に至った有害事象は本併用群2例(IAR)、対照薬群は1例(脳血管発作)、死亡に至った有害事象は認められなかった。
全体として、両投与群で31例に128件のIARが報告され、IARの発現割合は本併用群が24.7%(21/85例)、対照薬群が26.3%(10/38例)であった。
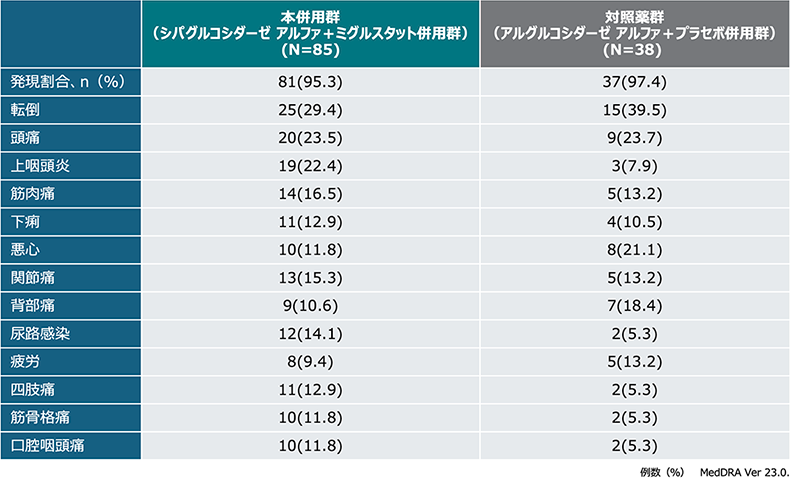
主な有害事象(いずれかの投与群で発現割合10%以上:123例)